糖尿病肾病研究中的FAS脂肪酸合成酶活性检测——当脂肪酸合成和氧化同时失调,意味着什么
糖尿病肾病(Diabetic Kidney Disease,DKD)是糖尿病最严重的微血管并发症之一,也是全球慢性肾脏病和终末期肾病最主要的病因。在中国,随着糖尿病患病率的持续攀升,DKD的疾病负担已经达到了相当严峻的规模。
但DKD的研究和治疗,长期以来面临一个困境:现有的一线治疗手段——包括血糖控制、血压管理、RAAS抑制剂和近年来兴起的SGLT2抑制剂——能够延缓肾功能恶化,但无法逆转已经发生的肾脏损伤,也不能阻止相当一部分患者最终进展到终末期肾病。理解DKD的发病机制,寻找新的治疗靶点,一直是这个领域的核心任务。
在这个背景下,肾小管的能量代谢异常越来越受到研究者的重视。肾脏近端小管上皮细胞是整个肾脏里代谢需求最高的细胞类型,它们的工作量极其繁重——重吸收约70%的肾小球滤液,主动转运大量溶质,这些都需要持续稳定的能量供应。而这种能量供应,在正常状态下主要依赖脂肪酸氧化(Fatty Acid Oxidation,FAO)而不是葡萄糖代谢。
当DKD发生时,这套精密的能量供应体系开始崩溃。脂肪酸氧化受阻,脂质在肾小管细胞内异常积累,线粒体功能障碍,氧化应激加剧,最终驱动肾小管纤维化和肾功能的进行性下降。在这条病理链条上,FAS活性检测能告诉我们什么?它在DKD研究里扮演的是什么角色?
肾小管细胞的能量代谢:为什么脂肪酸是主角
要理解FAS活性在DKD研究里的位置,需要先搞清楚肾小管细胞的代谢特点,因为它和大多数其他细胞类型有很大的不同。
大多数组织细胞在能量代谢上是"杂食性"的,葡萄糖、脂肪酸、氨基酸都能作为能量来源,根据底物可用性灵活切换。肾脏近端小管细胞则高度依赖脂肪酸氧化作为主要的ATP来源,这种依赖程度在正常生理状态下是非常突出的。原因在于近端小管细胞几乎不储存糖原,葡萄糖摄取能力有限,而FAO产生的ATP产率远高于糖酵解,更适合支撑近端小管高强度的主动转运工作。
这种能量代谢特点决定了一个关键的病理逻辑:一旦FAO受损,近端小管细胞就会陷入能量危机。这不是一个可以轻易被其他代谢通路补偿的问题,因为这些细胞本来就不擅长依靠葡萄糖产能,切换到糖酵解只能提供很少的能量,远不足以维持正常功能。能量危机导致主动转运功能下降,细胞内脂质积累,线粒体进一步受损,形成恶性循环。
在这个背景下,FAS(脂肪酸合成酶)在肾小管细胞里的角色就显得有些特殊。正常状态下,肾小管细胞并不是脂肪酸合成的主要场所——它们消耗脂肪酸,而不是大量合成。但在代谢失调的病理状态下,合成和氧化这两条通路的平衡被同时打破,FAO下降和FAS活性变化往往同时出现,共同构成了对肾小管代谢状态的完整描述。
CPT1A乙酰化:一个把FAO和FAS活性同时拉下去的修饰
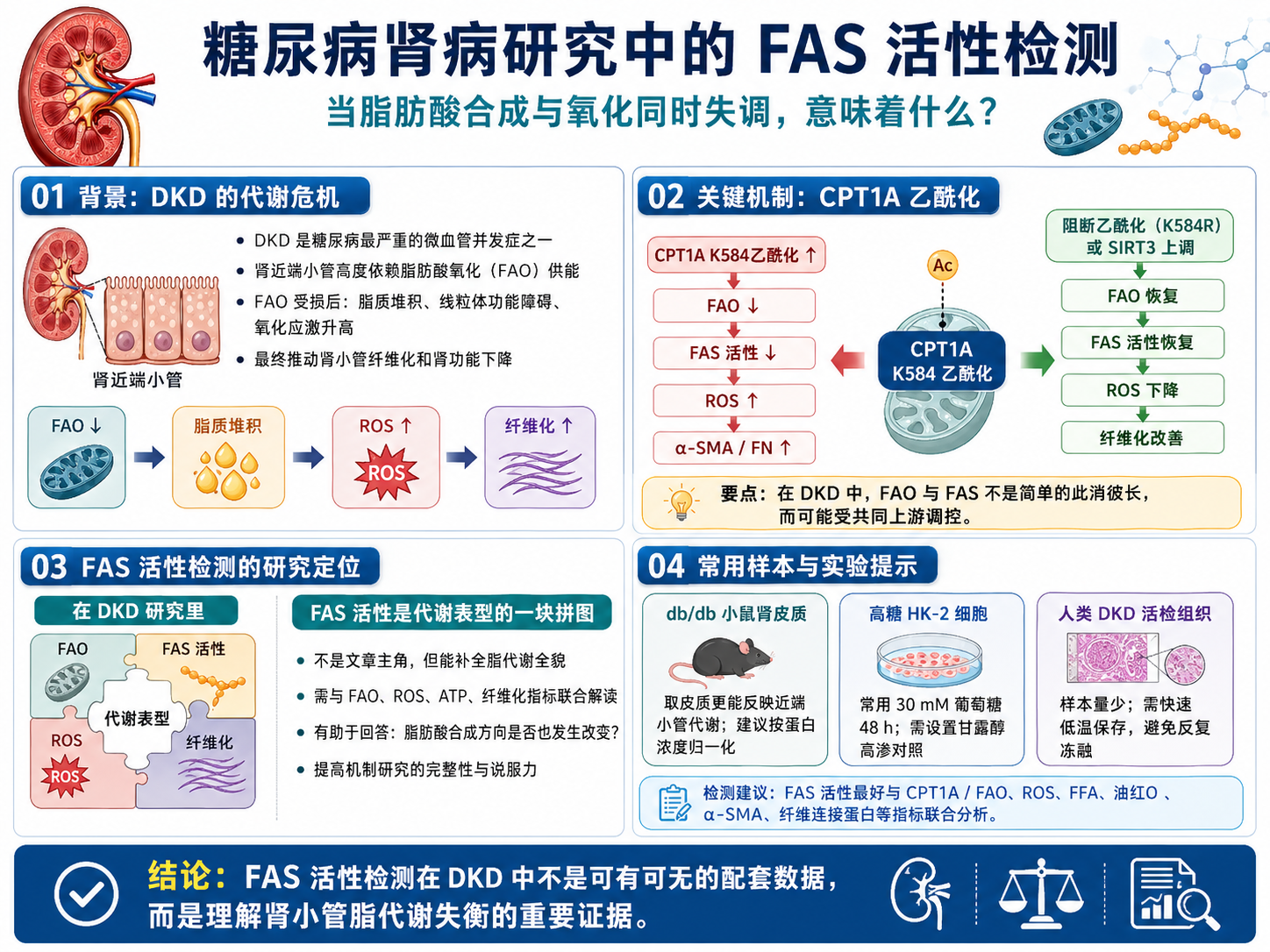
近期发表的一项DKD机制研究,把视角聚焦在了CPT1A(肉碱棕榈酰转移酶1A)的乙酰化修饰上,揭示了一个让人意外的发现——在DKD状态下,一个关键的翻译后修饰,同时影响了脂肪酸氧化和脂肪酸合成两条通路。
CPT1A是FAO通路的限速酶,位于线粒体外膜,负责把长链脂酰CoA转运进入线粒体基质,这一步是启动FAO的关键门控。没有CPT1A的转运,脂肪酸就进不了线粒体,FAO就无从谈起。
这项研究建立了db/db小鼠DKD模型,进行了全蛋白组乙酰化谱分析。结果发现,在DKD状态下,全局蛋白高乙酰化是一个普遍现象,而CPT1A是其中乙酰化程度最为显著的线粒体蛋白。研究进一步锁定了关键的乙酰化位点:赖氨酸584(K584)。
K584的乙酰化如何影响CPT1A的功能?乙酰化修饰改变了K584位点的电荷状态,影响了CPT1A的构象和催化效率,导致FAO显著下降。这一点不难理解——限速酶的活性受损,整条通路的通量自然下降。
但令人意外的是,同时检测FAS活性的结果显示,在CPT1A高乙酰化的DKD状态下,FAS活性也出现了显著下降,而不是代偿性升高。这打破了一个直觉上的预期——既然FAO下了,脂质消耗减少了,脂肪酸合成应该也相应下调,这听起来有道理。但DKD里观察到的FAS活性下降,背后的机制并不这么简单。
更关键的是,当研究者通过慢病毒介导的K584R点突变来阻断CPT1A在K584位点的乙酰化,观察到的结果是:FAO恢复,FAS活性同时恢复,ROS水平下降,肾纤维化标志物(α-SMA、纤维连接蛋白)也同步改善。FAO和FAS活性在这个实验设计里是同向变化的——两者一起下去,又一起回来。
这个发现告诉我们,在DKD的肾小管代谢紊乱里,FAO和FAS并不是简单的"氧化-合成"跷跷板关系。它们的失调可能有共同的上游调控机制,CPT1A的乙酰化状态以某种方式同时影响了两条通路的活性。理解这个关联,需要同时检测FAO相关指标和FAS活性,而不能只看其中一个。
FAS活性在DKD研究里的定位:代谢表型的一块拼图
和DLBCL研究里FAS作为治疗靶点的角色不同,在DKD的研究框架里,FAS活性检测扮演的是代谢表型描述的角色。
这个区别很重要,因为它决定了FAS活性数据在文章里的位置和解读方式。
在DLBCL里,研究者关心的问题是"FAS活性升高了多少,这种升高是否驱动了肿瘤细胞的增殖",FAS是因变量,也是潜在的干预靶点。在DKD的CPT1A乙酰化研究里,研究者关心的核心问题是"CPT1A乙酰化如何影响肾小管的代谢功能和纤维化进程",FAS活性是帮助回答这个问题的配套数据——它不是主角,但它的存在让代谢表型描述更完整,也让CPT1A乙酰化影响范围的判断更准确。
具体来说,在这类研究设计里,FAS活性数据通常和以下几组数据一起出现:
线粒体功能指标(线粒体膜电位、ROS水平、ATP产量)描述了线粒体整体的功能状态;FAO相关指标(CPT1A活性或表达、脂肪酸氧化速率)描述了脂质消耗方向的情况;纤维化标志物(α-SMA、纤维连接蛋白、胶原沉积)描述了病理损伤的程度;FAS活性则从脂质合成方向补充了代谢表型的另一面。这几组数据放在一起,才能构成对DKD肾小管代谢失调的立体描述,告诉读者"在这个病理状态下,合成和氧化两个方向的脂肪酸代谢同时出现了什么变化"。
从审稿的角度来看,一篇关注代谢机制的DKD文章,如果只报告了FAO相关数据而没有FAS数据,审稿人可能会问:脂肪酸合成方向有没有变化?两个方向的代谢失调是独立的还是相关的?补充FAS活性数据,能够预防这类质疑,让代谢表型描述没有明显的缺口。
DKD研究的样本类型和前处理
在DKD研究里,用于FAS活性检测的样本类型主要是以下几种,每种都有各自的特点和操作要点。
db/db小鼠肾脏组织
db/db小鼠是DKD研究里最经典的动物模型,由于瘦素受体基因突变导致肥胖、高血糖和胰岛素抵抗,表现出与人类DKD高度相似的肾脏病理特征,包括肾小球硬化、肾小管间质纤维化和蛋白尿。
从db/db小鼠肾脏取样做FAS活性检测,通常取约0.1 g肾皮质(而不是整个肾脏,因为皮质富含近端小管,是DKD病理改变最集中的部位),加入1 mL Extraction Buffer,冰浴匀浆,12000 g、4℃离心40分钟取上清。
需要注意的是,DKD小鼠肾脏可能有脂质积累,虽然不像肝脏那样会形成明显的脂肪层,但离心后如果上层有轻微的浑浊,仍然要小心吸取上清,避免混入脂质颗粒。另外,DKD状态下肾脏的蛋白总量可能因为纤维化的存在而与正常对照组有所不同,这使得蛋白浓度归一化在DKD组织样本里尤其重要——不做归一化直接比较FAS活性,可能把纤维化带来的蛋白组成变化混淆为代谢活性的变化。
取样时间点的选择也值得注意。DKD是一个进展性疾病,早期(比如db/db小鼠8-12周)和晚期(20周以上)肾脏的代谢表型差异很大。实验设计里需要明确取样的周龄,如果要研究疾病进展过程中FAS活性的动态变化,需要设置多个时间点,这对样本量的需求会相应增加。
高糖处理的肾小管上皮细胞
体外细胞模型是DKD研究里另一个重要的实验系统。HK-2(人近端小管上皮细胞系)是最常用的选择,用30 mM葡萄糖处理48小时来模拟DKD的高糖微环境,是这篇CPT1A乙酰化研究里使用的体外模型。
HK-2细胞是贴壁细胞,收集前需要胰酶消化,消化要充分,否则细胞收集率偏低,后续的酶活检测信号会偏弱。收集约5×10⁶个细胞,预冷PBS清洗两次,800 g离心2分钟弃上清,加入1 mL Extraction Buffer超声裂解,之后12000 g、4℃离心40分钟取上清。
高糖处理的HK-2细胞在代谢状态上和正常培养的细胞有显著差异,FAS活性的变化幅度可能比动物组织样本更容易观察到,因为细胞系实验的个体变异更小,组间对比更干净。但也因为这个原因,细胞实验的数据需要在动物实验里得到验证,才能支撑更强的机制性结论。
高渗透压对照是体外高糖实验里经常被要求设置的对照组——用甘露醇维持和高糖组相同的渗透压,但不提供额外的葡萄糖代谢底物,用来排除高渗透压本身(而非高糖代谢)对细胞代谢的影响。如果高糖组和甘露醇对照组之间FAS活性有显著差异,说明观察到的变化来自葡萄糖代谢的改变,而不只是渗透压效应。这个对照组的设置不是可选项,是高糖体外实验方法学严谨性的基本要求。
人类DKD肾脏活检组织
临床样本是最有说服力的,但也是最难获取的。如果有条件获得DKD患者的肾脏活检组织,FAS活性检测能够把动物实验和细胞实验的发现直接延伸到人类疾病层面。
活检组织通常量很少,可能只有几十毫克,这对检测来说是一个挑战。需要根据实际取样量等比例调整Extraction Buffer的用量,确保裂解效率,同时上样量也可能需要从20 µL调整到更少,然后在数据计算时进行相应的换算。使用亚科因生物KTB2240的在线计算工具时,确保输入的样本质量或蛋白浓度参数与实际情况一致。
临床样本还需要注意保存条件。新鲜组织如果无法立即检测,应立刻在液氮里速冻后转移到-80℃保存,不建议在普通-20℃长期保存,酶活性在-20℃下的保存稳定性远不如-80℃。每次解冻后必须当天完成检测,反复冻融对FAS活性的破坏是不可逆的。
在CPT1A乙酰化研究框架里,FAS活性数据如何设计和解读
基于这篇研究的实验逻辑,FAS活性检测通常出现在以下几个实验场景里。
场景一:验证DKD模型的代谢表型
在建立好DKD动物模型(db/db小鼠)或细胞模型(高糖处理HK-2)之后,首先需要确认模型本身的代谢表型是符合预期的。除了常规的血糖、体重、肾功能指标(血肌酐、尿蛋白),代谢层面需要检测FAO相关指标和FAS活性,确认在你的实验系统里,DKD状态确实导致了脂肪酸代谢的双向失调——既有FAO下降,也有FAS活性变化。
这一步建立表型的数据,是后续机制研究的前提。如果你的DKD模型里没有观察到FAS活性的变化,后面关于CPT1A乙酰化同时影响FAO和FAS的结论就失去了基础,需要先回头确认模型和取样是否有问题。
场景二:验证CPT1A乙酰化干预对FAS活性的影响
这是最核心的实验设计。在K584R点突变(阻断乙酰化)的实验组里,预期FAO应该恢复,FAS活性也应该相应恢复。同样,如果使用SIRT3过表达来促进CPT1A去乙酰化(SIRT3是一种线粒体去乙酰化酶,在DKD中表达下调,是CPT1A乙酰化程度升高的原因之一),预期也应该观察到FAO和FAS活性的同步改善。
这组实验里设置的对比:野生型CPT1A(高乙酰化)vs K584R突变(低乙酰化),或者对照组(低SIRT3)vs SIRT3过表达组,FAS活性数据放在FAO数据旁边,共同构成"CPT1A乙酰化状态影响肾小管整体脂肪酸代谢"的证据。
如果两个方向(FAO和FAS)的变化都和乙酰化状态一致,说明CPT1A乙酰化的影响确实是广泛的,不只局限于FAO通路,对脂肪酸代谢的整体调控都有影响。如果只有FAO的变化而FAS没有,说明两者可能是独立调控的,需要重新审视实验假设。
场景三:联合其他代谢指标构建完整的代谢表型
FAS活性单独出现的时候,只能回答"脂肪酸合成方向有没有变化"这一个问题。在DKD研究里,要构建完整的代谢表型,通常还需要配合以下指标:
线粒体膜电位(JC-1染色)和ROS水平(DCFH-DA染色)反映线粒体功能状态;CPT1A蛋白表达量(Western blot)和乙酰化水平(免疫沉淀后Western blot)说明CPT1A的修饰状态;游离脂肪酸(FFA)含量反映细胞内脂质积累程度;油红O染色直观呈现脂滴的分布和大小;α-SMA、纤维连接蛋白的蛋白表达量描述纤维化程度。
FAS活性在这个多指标体系里处于"脂肪酸代谢合成方向"的位置,和FAO相关指标形成一对,共同描述脂肪酸代谢的双向状态。这种多指标配合的数据呈现方式,在代谢机制文章里是相对成熟的范式,也是审稿人比较习惯看到的数据组合。亚科因生物有配套的游离脂肪酸检测试剂盒(KTB2230)和总胆固醇检测试剂盒(KTB2220),可以和KTB2240在同一批样本上同步检测,减少批次差异。
关于FAS活性在DKD里的方向:升高还是降低
这是一个需要在实验前想清楚的问题,因为DKD里FAS活性的变化方向,和直觉预期之间有时候存在落差。
从直觉上来说,既然DKD里FAO受损,脂肪酸消耗减少,那脂肪酸合成是不是应该代偿性升高?这个推断听起来有逻辑,但CPT1A乙酰化研究里观察到的是相反的结果——FAS活性和FAO是同向下降的。
这个"同向下降"背后的机制目前还不完全清楚。一种可能的解释是,在严重的线粒体功能障碍状态下,细胞整体的代谢活跃程度都下降,包括需要消耗大量ATP和NADPH的脂肪酸合成通路。另一种可能是,CPT1A乙酰化通过某种尚未完全阐明的信号机制,影响了FAS的活性或者表达。也可能是两者都有。
这意味着在你设计实验的时候,不要预设FAS活性一定是升高的。不同的DKD模型、不同的疾病阶段、不同的干预手段,FAS活性的变化方向可能不一样。预实验在这里尤其重要——先在2-3个预期差异最大的样本上做一轮,看清楚ΔA的大致范围和变化方向,再决定正式实验的样本量和统计设计。
从检测操作的角度来说,FAS活性下降意味着ΔA值会偏小,如果下降幅度很大,ΔA可能接近检测下限(0.0005)。这种情况下,可以适当增加上样量,从20 µL增加到25或30 µL,或者增加裂解时的样本取量,提高上清里的蛋白浓度,让信号更容易被检测到。
DKD研究里容易被忽视的几个方法学细节
肾皮质和肾髓质要分开取
肾脏不同区域的细胞类型和代谢特点差异很大。近端小管主要分布在皮质,是DKD病理改变最集中的区域,也是FAO最活跃的区域。如果取样时把皮质和髓质混在一起匀浆,会稀释来自皮质近端小管细胞的信号,让FAS活性的组间差异变小,难以检测到真实的变化。建议在解剖时用手术刀划开肾脏,在体视镜下分离皮质,专门取皮质组织用于代谢检测。
蛋白归一化在DKD组织里特别重要
前面提到过,DKD肾脏因为纤维化的存在,细胞外基质蛋白(胶原等)的比例增加,而功能性上皮细胞减少。如果按组织质量归一化(U/g鲜重),同样1 g组织里,DKD肾脏含有的功能细胞比正常肾脏少,这个差异本身就会导致FAS活性表观上"降低",即使每个细胞的FAS活性其实没有变化。按蛋白浓度归一化(U/mg prot)能够在一定程度上消除这种混淆,是DKD组织样本里更推荐的归一化方式。
但这里还有一个更深层的问题:如果纤维化本身导致总蛋白里细胞外基质蛋白比例上升、功能细胞蛋白比例下降,按总蛋白归一化也不能完全消除这种偏差。更严格的做法是同时报告按质量和按蛋白归一化的两套数据,或者使用细胞类型特异性标志物(比如近端小管标志物AQP1)来对FAS活性做更精准的归一化。这种做法在方法学要求比较高的情况下才需要,大多数情况下按总蛋白归一化已经足够。
高糖体外实验的处理时间点选择
30 mM高糖处理HK-2细胞,不同的处理时间(24小时、48小时、72小时)对代谢表型的影响不同。CPT1A乙酰化研究使用的是48小时,这是文献里比较常见的时间点,在这个时间点上细胞通常已经建立了稳定的高糖代谢应激状态,但还没有出现大规模的细胞死亡,检测到的FAS活性反映的是存活细胞的代谢状态。如果处理时间太长,细胞活力下降明显,检测到的代谢活性变化里会混入细胞死亡的因素,结果解读会变得复杂。
这个研究方向的上下游延伸
理解了CPT1A乙酰化和FAS活性之间的关联,自然会引出几个值得继续探索的问题,对于正在设计研究方向的人来说是值得考虑的延伸点。
上游:是谁在调控CPT1A的乙酰化状态
乙酰化是可逆修饰,受到乙酰化酶(如各种KAT家族成员)和去乙酰化酶的动态调控。在DKD的背景下,SIRT3(一种线粒体去乙酰化酶,属于Sirtuin家族)的表达下调是CPT1A高乙酰化的重要原因之一,这已经有文献支撑。那么是什么导致了DKD里SIRT3的下调?高糖、脂毒性、氧化应激都可能参与其中,但具体的调控链条还有待厘清。另外,除了SIRT3,是否还有其他去乙酰化酶参与了CPT1A的去乙酰化调控,在不同的细胞区室里是否有分工?这些都是可以展开的研究问题。
下游:FAS活性和CPT1A乙酰化之间的机制连接是什么
这是最直接的未解问题。CPT1A是FAO的限速酶,它的乙酰化怎么会影响到FAS(脂肪酸合成酶)的活性?两者之间的机制连接目前还不清楚。一种可能是通过Acetyl-CoA/Malonyl-CoA的代谢交叉点——FAO和FAS在这个节点上共享底物,FAO受损可能影响Acetyl-CoA的代谢流向,进而影响FAS的底物供给和活性。另一种可能是通过更广泛的代谢信号(比如AMPK通路),FAO下降引发的能量应激激活AMPK,而AMPK能够磷酸化并抑制乙酰CoA羧化酶(ACC),减少Malonyl-CoA的供应,从而抑制FAS的活性。
如果能够阐明这个机制连接,这将是一个有重要发表价值的发现,因为它揭示了脂肪酸代谢里合成和氧化两条通路之间的协调机制,不只是DKD领域,对代谢生物学的理解也有普遍意义。
但在机制问题还没有答案之前,有一个实际的推论值得重视:正因为FAO和FAS同向变化的机制尚不清楚,FAS活性检测在DKD研究里就不是可有可无的配套数据,而是必要的表型证据。 如果你的研究只测了FAO相关指标,而没有同时测FAS活性,你就无法知道在你的实验体系里这两条通路是否同向变化,也就无法判断你观察到的代谢失调是CPT1A乙酰化这篇文献里描述的那种模式,还是另一种完全不同的模式。表型数据越完整,机制假说才越有根基。这不是为了凑数据,而是因为在机制未明的情况下,完整的表型描述本身就是最有价值的科学贡献之一。
横向:其他肾脏疾病中的FAS活性
DKD是肾脏脂代谢研究里最受关注的疾病模型,但类似的代谢异常在其他肾脏疾病里也可能存在,包括局灶节段性肾小球硬化(FSGS)、IgA肾病、急性肾损伤(AKI)等。如果你的研究已经建立了DKD里的FAS活性检测体系,横向扩展到其他肾脏疾病模型,成本增加有限,但可能发现新的代谢表型关联,为后续的比较研究提供基础。
这个研究方向需要客观面对的局限
在对DKD脂代谢研究保持热情的同时,有几个现实层面的局限需要清醒认识。
CPT1A乙酰化和FAS活性的因果关系尚不完全确立
目前的数据显示两者存在关联性变化,K584R突变的实验也提供了干预证据,但CPT1A乙酰化究竟是通过什么机制影响FAS活性,还没有直接的机制证据。在这个问题没有回答清楚之前,把CPT1A乙酰化作为调控FAS活性的上游机制来描述,在科学上是需要谨慎的。这个局限在写作和文章讨论部分需要如实呈现。
动物模型到人类DKD的外推有距离
db/db小鼠是2型糖尿病的遗传模型,和人类DKD的发病过程在机制上有相似之处,但也有重要差异。人类DKD的病程更长,影响因素更复杂(高血压、血脂异常、肥胖往往同时存在),而且不同患者的DKD亚型在代谢表型上可能有显著差异。db/db小鼠里观察到的FAS活性变化,在人类DKD患者样本里是否同样存在,需要有临床样本的数据来支撑,不能简单外推。
FAS活性作为DKD生物标志物的可行性仍需验证
FAS活性检测目前的平台是基于组织或细胞裂解液,对于临床应用来说,有创的组织活检限制了它作为常规监测指标的可行性。血清样本虽然可以直接检测,但血清FAS活性和肾脏局部FAS活性之间的对应关系,目前还缺乏系统性的验证数据。
在论文方法部分如何描述
使用KTB2240(亚科因生物,CheKine™脂肪酸合成酶活性检测试剂盒,微量法)进行FAS活性检测,方法部分可以参考以下描述:
肾脏组织FAS活性按照试剂盒说明书(CheKine™ Micro Fatty Acid Synthetase Activity Assay Kit,KTB2240,Abbkine Scientific)进行检测。取约0.1 g肾皮质组织,加入1 mL提取液冰浴匀浆,12000 g、4℃离心40分钟取上清。每孔检测体系包含20 µL样本上清和180 µL工作液(含NADPH、乙酰CoA和丙二酰CoA),在96孔UV板中37℃孵育后,于340 nm处连续检测吸光度,记录第10秒(A₁)和第70秒(A₂)的吸光度值,以ΔA=A₁-A₂计算FAS活性。结果以U/mg蛋白表示,蛋白浓度采用BCA法(KTD3001,Abbkine Scientific)测定。
对于细胞样本,将"取约0.1 g肾皮质组织,加入1 mL提取液冰浴匀浆"替换为"收集5×10⁶个细胞,预冷PBS清洗后加入1 mL提取液超声裂解",其余步骤相同。
写在最后
DKD的代谢研究正在经历一个从"描述现象"到"理解机制"的转变。早期的研究确认了脂肪酸代谢紊乱在DKD里的存在,现在的研究开始追问:是什么在驱动这种紊乱,合成和氧化两个方向的失调之间有什么关系,能不能通过干预代谢修饰来逆转这种紊乱。
CPT1A乙酰化研究里同时检测FAO和FAS活性,不只是数据上的全面,更反映了一种研究思路——肾小管的代谢健康,需要从合成和氧化两个方向同时来评估。FAS活性检测在这里的价值,正是提供了这个视角里"合成方向"的那一半数据。
把这两半数据放在一起,才能看清楚在DKD的病理状态下,肾小管细胞的脂肪酸代谢是怎么失控的,以及当我们试图干预这种失控时,两个方向的代谢是否能够协调恢复。这是一个还没有完全解答的问题,也是这个研究方向仍然值得投入的原因。
