FAS脂肪酸合成酶活性为什么这样检测?NADPH消耗法的原理、优势与边界
研究脂代谢的人,绕不开FAS。
脂肪酸合成酶(Fatty Acid Synthase,FAS)是脂肪酸从头合成的核心催化酶,几乎参与了所有涉及脂质积累的生理和病理过程。肝脏里的脂肪合成、肿瘤细胞的代谢重编程、糖尿病状态下的脂质紊乱、神经炎症里的脂滴蓄积——课题只要碰到脂代谢表型,FAS通常是第一个需要回答的问题。
但"需要检测FAS"这件事,比很多人想的要复杂一点。FAS可以从蛋白表达量、基因转录水平、酶活性三个层面来描述,而这三个层面有时候并不一致。表达量高不等于活性强,活性强不等于产物就多。真正能反映FAS在样本里"到底在干多少活"的,是酶活性检测。
那么,FAS活性该怎么检测?目前最主流的方法是什么?它的原理是什么,能回答什么问题,又有哪些边界需要清楚?这篇文章试图把这些问题说清楚。
FAS是一个什么样的酶
在讨论检测方法之前,先要对FAS这个酶有基本的了解,否则很多方法选择的逻辑就难以理解。
哺乳动物的FAS是一个大型多功能酶,分子量约为270 kDa,以同源二聚体的形式发挥功能,整个二聚体接近540 kDa。它包含七个功能域,能够依次完成脂肪酸从头合成所需的全部催化步骤,从两碳的乙酰辅酶A(Acetyl CoA)出发,以丙二酰辅酶A(Malonyl CoA)为碳链延伸单元,每个延伸循环消耗两分子NADPH,最终生成十六碳的棕榈酸(palmitate)作为主要产物。
这个过程的总反应可以简写为:
Acetyl-CoA + 7 Malonyl-CoA + 14 NADPH + 14 H⁺ → Palmitate + 7 CO₂ + 14 NADP⁺ + 8 CoA + 6 H₂O
两个关键底物的消耗——丙二酰CoA和NADPH——都可以作为检测FAS活性的切入点。其中NADPH的检测更简便,这就是现代FAS活性检测方法的起点。
FAS在正常组织中的表达因部位不同而差异显著。肝脏、乳腺、脑、肾脏和脂肪组织表达丰富,是脂肪酸合成活跃的场所;骨骼肌表达相对较低。在病理状态下,尤其是多种肿瘤里,FAS的表达和活性会显著上调,这也是它成为肿瘤代谢研究热点的原因之一。
测"量"和测"活",是两件不同的事
在进入检测方法之前,有必要先把一个概念辨析清楚:检测FAS蛋白表达量,和检测FAS酶活性,回答的是完全不同的问题。
Western blot和免疫组化(IHC)是检测FAS蛋白量的经典手段,告诉你的是"这个样本里有多少FAS蛋白"。这个信息固然重要,但蛋白的量和酶的活性之间,有时候差距非常大。
FAS的活性受到多重调节——转录后修饰(比如乙酰化、磷酸化)、底物可用性、产物反馈抑制、辅因子状态,都会影响FAS实际的催化效率。有研究发现,FAS乙酰化修饰可以显著降低其催化活性,而这种变化在蛋白表达量上几乎看不出来。也就是说,Western blot显示FAS蛋白量没有变化,并不代表FAS活性没有变化;反之,FAS活性的差异,也未必会在蛋白水平上有所体现。
这正是为什么一篇好的脂代谢机制文章,往往同时报告FAS的表达量数据和活性数据——两者互补,共同支撑对FAS功能状态的完整描述。发表于Leukemia的研究探讨ZDHHC21/FASN轴对弥漫大B细胞淋巴瘤的影响时,FAS活性检测就是除蛋白层面分析之外的关键功能性证据之一。只有活性数据,才能直接回答"FAS在这个状态下到底有多活跃"。
FAS活性检测的几种思路
历史上,FAS活性检测走过几条不同的技术路线。
最早期的方法是放射性同位素标记法,用¹⁴C标记的乙酰CoA或丙二酰CoA作为底物,通过测定放入产物里的放射性来计算底物消耗量,进而推算FAS活性。这个方法灵敏度高,但需要放射性操作资质,废弃物处理麻烦,成本也高,现在在普通科研实验室里基本已经淡出使用。
另一条思路是直接检测产物。棕榈酸的生成量可以通过气相色谱或液相色谱质谱(LC-MS)来定量,精度很高,能区分不同链长的脂肪酸,但仪器要求和操作复杂度都不低,通常只在有专门代谢组学平台的实验室才可行,不适合作为常规的功能性检测手段。
还有一类思路是荧光法,用荧光标记的NADPH类似物作为底物,通过荧光信号的变化来检测NADPH消耗,灵敏度理论上比比色法更高,检测下限更低,适合处理极微量样本。但荧光检测对环境光控制要求严格,操作难度更大,试剂成本也更高,而且荧光标记底物的结构改变可能影响FAS对它的识别效率,导致测出来的活性和真实值之间有一定偏差。
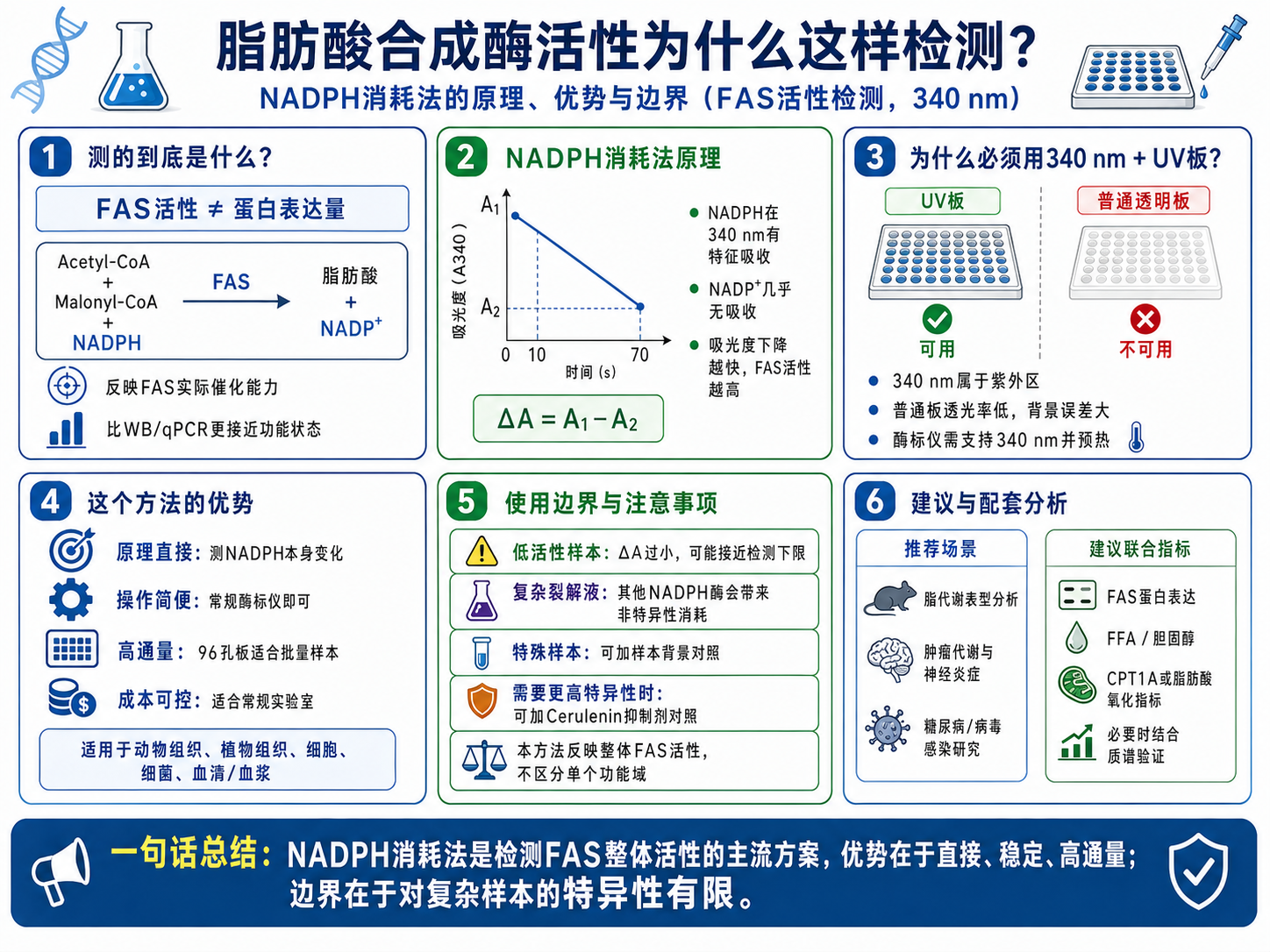
目前在科研实验室里使用最广泛的,是基于NADPH在340 nm吸收的分光光度法,也就是比色法。原因很简单:原理直接、操作简便、兼容常规酶标仪、成本可控、可以高通量操作。这种方法的检测灵敏度对大多数样本类型来说完全够用,数据的可重复性也经过了大量文献的验证。
NADPH消耗法:原理为什么是这样的
NADPH(烟酰胺腺嘌呤二核苷酸磷酸,还原型)和NADP⁺(氧化型)在分子结构上只差一对电子和一个氢离子,但这个差别带来了一个在实验上极为有用的光学特性:NADPH在340 nm处有强烈的特征吸收峰,而NADP⁺在这个波长几乎没有吸收。
这意味着,在一个含有FAS的反应体系里,只要加入底物(乙酰CoA、丙二酰CoA)和NADPH,随着反应进行,NADPH不断被消耗转化为NADP⁺,340 nm处的吸光度就会持续下降。下降的速率越快,说明NADPH被消耗得越快,FAS的催化活性越高。
这个逻辑是直接的,不需要引入任何间接的指示体系。测的就是底物本身的光学变化,中间没有多余的耦联反应,干扰来源少,信号解读也直观。
从检测灵敏度的角度来看,NADPH的摩尔消光系数在340 nm处是6.22×10³ L/mol/cm,这个数值不算很高(相比之下,荧光法的灵敏度通常高出1-2个数量级),但对于动物组织、细胞裂解液、血清等常见样本类型,FAS活性通常在可检测范围内,不需要过度担心灵敏度不够的问题。
为什么是340 nm,这个波长有什么特别的
340 nm处于紫外光区域,这也是为什么检测FAS活性必须用UV板而不能用普通透明酶标板的原因——普通聚苯乙烯在这个波段的透过率极低,不同孔之间的背景吸光度差异可以大到完全掩盖来自NADPH消耗的信号变化。UV板(紫外透明板)使用的材料在340 nm处透过率高且均匀,是做这类检测的唯一合适选择。
340 nm同时也是很多其他需要检测NADH或NADPH变化的酶促反应的通用检测波长,这意味着这个波长在酶学研究领域积累了大量的方法学验证和参考数据,测出来的数值在文献中有充分的横向对比基础。
有一点需要注意:某些样本本身在340 nm处会有背景吸光度,或者含有会干扰读数的内源性物质。严重溶血的血清、含有大量还原性物质的植物提取物,在这个波段的背景值可能不可忽略。处理这类样本时,需要额外设置样本背景对照(用不含底物的反应液代替工作液,同样跑一遍ΔA,从实验孔的ΔA中扣除),才能得到真实反映FAS活性的数值。这个操作步骤在一般说明书里不会特别强调,但在遇到高背景样本时是必要的。
微量法是什么,和传统分光光度计法有什么区别
早期的NADPH消耗法通常在比色皿里操作,用台式分光光度计读数,单次检测体积在1-3 mL左右,每次只能测一个样本,操作效率低。这种格式对于样本量少、只需要测几个数据点的场景还能接受,但面对需要同时处理几十个样本的实验设计,就力不从心了。
微量法是这个方法在通量上的重要升级。把反应体系缩小到96孔板的格式,每孔只需要200 µL,一块板可以同时跑几十个样本,加上多通道移液器的配合,整体效率提升非常显著。亚科因生物的KTB2240就是基于这个格式设计的——96孔UV板,每孔200 µL反应体系(20 µL样本+180 µL工作液),在酶标仪上读340 nm吸光度。
从数据质量的角度来看,微量法和传统分光光度计法得到的结果是可比的,只要光径参数在计算时正确代入就行。96孔UV板的标准光径是0.5 cm,比比色皿的1 cm短,在用Beer-Lambert定律折算浓度时需要注意。KTB2240说明书里给出的计算公式已经把0.5 cm的光径参数折进综合常数里了,直接套用公式不需要另行换算,但如果你改变了板子规格(比如改用384孔板),就必须重新核对光径和反应体积对应的综合常数。
这个方法测的是整体FAS活性,还是某个具体步骤
这是一个值得深入理解的问题,和实验结果的解读直接相关。
NADPH消耗法测的是FAS催化整个脂肪酸合成反应过程中的总NADPH消耗速率,反映的是FAS作为整体的催化效率,而不是某个单独功能域(比如烯酰还原酶域或酮酰还原酶域)的活性。这意味着,如果某个处理使FAS的某个功能域受到影响,但不影响整体的NADPH消耗速率,这个方法可能捕捉不到这种变化。
但在绝大多数生理和病理研究场景里,研究者关心的是"样本里FAS的整体合成能力有没有变化",而不是某个特定催化步骤是否受到影响。从这个角度来看,NADPH消耗法提供的整体活性数据,正好对应了最常见的研究问题,是合适的。
如果研究目的是探讨FAS某个特定功能域的活性变化,或者需要区分不同的NADPH消耗来源(因为细胞裂解液里除了FAS,还有其他依赖NADPH的酶),就需要更精细的方法,比如使用特异性底物、加入FAS抑制剂(如Cerulenin或C75)做对照实验来扣除非特异性NADPH消耗,或者借助更高分辨率的质谱方法。这些是方法的边界,在设计实验时需要有清醒的认识。
反应时间窗口为什么是60秒
NADPH消耗法的标准操作是在加入样本后,记录第10秒和第70秒时的吸光度,用这60秒时间窗内的ΔA来计算FAS活性。这个时间设置不是随意的。
加入样本之后,FAS催化反应并不是瞬间达到最大速率的。有一个短暂的启动阶段,酶和底物需要接触、结合、完成第一个催化循环,反应速率才会达到稳定的最大值。这就是为什么从第10秒而不是从0秒开始记录——前几秒的信号变化不在线性阶段,用于计算会带来误差。
另一方面,随着反应进行,底物浓度逐渐下降,当NADPH浓度降到一定程度,反应速率开始随浓度下降而降低,偏离初始的线性关系。使用过长的时间窗口,测到的就不再是最大反应速率,而是一个混合阶段的平均值,计算出来的酶活会偏低。
60秒的窗口(10秒到70秒)是在这两个边界之间找到的一个合理平衡:既跳过了启动阶段,又没有延伸到底物耗尽开始影响速率的区间,保证了计算所用的ΔA对应的是反应的线性阶段,测出来的才是真实的初速度,也就是最大酶活。
这同时也解释了为什么工作液要在37℃预孵育15分钟:预孵育让底物在加入样本前就已经处于活化状态,加入含FAS的样本后,反应能更快进入稳定的线性阶段,10秒到70秒这个窗口就能更准确地捕捉到初速度。省掉预孵育,反应启动延迟,第10秒可能还没进入线性阶段,测出来的ΔA偏低,计算出来的酶活就会失真。
底物的选择和浓度对结果的影响
KTB2240的工作液中包含三种关键成分:NADPH、乙酰CoA和丙二酰CoA,分别对应FAS反应的还原力来源、链起始单元和链延伸单元。三者的浓度比例是经过优化的,保证在正常的FAS活性范围内,底物浓度足以维持酶在最大速率下工作,NADPH的消耗速率真实反映FAS活性,而不是被底物限制。
这也解释了工作液配制为什么要严格按照说明书的比例来:NADPH 16 µL、Acetyl CoA 4 µL、Malonyl CoA 8 µL、Assay Buffer 152 µL,不是可以随意调整的。如果NADPH加少了,底物浓度不足,反应速率会被NADPH浓度限制,测出来的ΔA偏低,酶活被低估。如果底物浓度过高,在样本FAS活性很低的情况下可能没什么问题,但在FAS活性很高的样本里,60秒内NADPH消耗过多,ΔA值超出线性范围,同样会导致计算误差。
从这个角度来理解,ΔA值的合理范围(0.0005到0.4之间)其实是对应了底物浓度能够维持线性反应速率的窗口。ΔA超出这个范围,不管是太小还是太大,都意味着检测条件偏离了设计前提,需要调整样本量或者稀释倍数,让ΔA回到线性区间,数据才是可用的。
这个方法在文献里是怎么被使用的
一个检测方法在文献里被引用的方式,能反映出这个方法实际上在解决什么样的科学问题。
在发表于Leukemia的弥漫大B细胞淋巴瘤研究中,FAS活性检测被用来验证ZDHHC21对FASN(FAS的编码基因)功能的调控效应——光有蛋白表达量的数据不够,还需要活性数据来证明这种调控在功能层面上是真实发生的。这是FAS活性检测在肿瘤脂代谢靶点研究里最典型的使用逻辑:作为功能性证据,和蛋白层面的数据互相支撑。
在创伤性脑损伤(TBI)相关的研究中,FAS乙酰化被发现能影响小胶质细胞的脂滴蓄积和促炎活性,FAS活性检测在这里的作用是直接证明乙酰化修饰对酶催化效率的影响。这个场景里,蛋白总量没有变,修饰状态变了,活性变了,Western blot完全看不出来,必须靠酶活数据来说话。
在糖尿病肾病的代谢研究中,FAS活性是脂代谢表型分析的一部分,和CPT1A介导的脂肪酸氧化、线粒体功能一起构成了对肾小管代谢状态的综合描述。FAS活性高说明从头合成活跃,结合氧化代谢的数据,可以推断脂质在肾小管细胞里是否发生了异常积累。
在PLoS Pathogens发表的HSV-1感染研究中,FAS活性被用来描述病毒感染如何改变宿主细胞的脂质代谢环境——病毒复制需要脂质,劫持宿主的FAS是一种已经在多种病毒中观察到的策略,活性数据在这里直接反映了这种代谢劫持的程度。
这些案例里有一个共同的逻辑:FAS活性检测提供的是蛋白层面和转录层面无法替代的功能性证据,在机制研究里扮演的是"证明功能真实发生"的角色,而不只是"描述表达量高低"。
这个方法的边界在哪里
任何方法都有边界,知道边界是正确使用方法的前提。
灵敏度边界。 对于FAS活性极低的样本,比如某些静止期细胞或者FAS本来表达就少的组织,ΔA可能非常小,接近检测下限。这种情况下,增加上样量是一个选择,但受限于工作液体系的线性范围,增加的空间有限。如果对灵敏度有极高要求,荧光法或者基于质谱的检测方法在这个场景下会是更合适的选择。
特异性边界。 细胞裂解液里不只有FAS在消耗NADPH。其他依赖NADPH的脱氢酶、还原酶也会贡献一定的NADPH消耗,这些非特异性消耗会叠加在FAS催化的信号上,让测出来的酶活偏高。在大多数样本里,FAS贡献的NADPH消耗占主导,这种偏差在可接受范围内。但这里需要说一个容易被低估的问题:在复杂样本体系里,比如细胞裂解液,除了FAS之外还有大量其他依赖NADPH的酶——各种脱氢酶、还原酶——它们同样会消耗NADPH,同样会让340 nm的吸光度下降。单纯设置"无底物对照"(用不含三种底物的反应液代替工作液)只能扣除样本本身的静态背景吸光度,但扣不掉那些在加入底物之后被激活或受影响的非特异性NADPH消耗。
在这种情况下,更严谨的做法是引入FAS特异性抑制剂对照。Cerulenin是目前最常用的FAS特异性抑制剂,它共价结合FAS的β-酮脂酰合酶功能域,选择性阻断FAS的催化活性。具体操作是:设置一组加入Cerulenin预处理的平行样本,同样加入工作液,读取ΔA。这个孔里剩余的NADPH消耗来自非FAS的其他酶,是真实的非特异性背景。用总ΔA减去这个背景值,得到的才是严格意义上的FAS特异性NADPH消耗,对应的酶活数据才是可靠的。
这个做法在方法学要求较高的研究里,特别是需要在审稿时证明检测特异性的场景下,是值得认真考虑的。对于常规的组织或细胞样本,如果不涉及对FAS特异性有特别质疑的场景,通常无底物空白对照已经够用。但如果你的样本来源特殊、内源NADPH酶活复杂,或者数据出来之后在逻辑上难以自洽,Cerulenin抑制剂对照是第一个应该想到的补救方案。
样本类型边界。 这个方法已经在动物组织、植物组织、细胞、细菌和血清血浆五类样本中得到验证,覆盖了绝大多数科研场景。但对于某些特殊来源的样本——比如含有大量色素的植物提取物、脂质含量极高的脂肪组织提取物——背景干扰可能比较显著,需要通过设置样本背景对照来修正。
功能域特异性边界。 如前所述,这个方法测的是FAS的整体催化效率,不区分各功能域的贡献。如果研究问题涉及FAS特定功能域的调控,需要结合其他方法。
和其他脂代谢检测指标的配合使用
FAS活性数据单独出现的时候,能回答"FAS有多活跃"这个问题。但在一个完整的脂代谢研究框架里,它通常需要和其他指标配合,才能构成对脂代谢状态的完整描述。
游离脂肪酸(FFA)含量反映的是细胞或组织里游离脂肪酸的实际积累量,是FAS合成活动的下游结果之一。如果FAS活性高,但FFA含量没有相应增加,说明合成出来的脂肪酸被快速利用或者转运走了。如果FFA高但FAS活性不高,说明FFA的积累更多来自外源摄入或者储存脂肪的释放,而不是从头合成。两个指标结合,才能区分这些不同的代谢来源。
胆固醇(包括总胆固醇和游离胆固醇)是另一个常见的配套检测指标。脂肪酸和胆固醇是脂质代谢里的两条主线,一起检测有助于描述更完整的脂代谢表型。
对于关注线粒体功能的研究,FAS活性(代表合成方向)和CPT1A活性(代表氧化方向)的组合是一个常见的配对——合成和氧化的平衡决定了细胞内脂质是积累还是消耗,这种平衡在代谢性疾病(比如糖尿病肾病、非酒精性脂肪肝)的发病机制里是一个核心问题。
亚科因生物在这几个方向上有配套产品,包括游离脂肪酸检测试剂盒(KTB2230)、游离胆固醇检测试剂盒(KTB2210)、总胆固醇检测试剂盒(KTB2220),可以和KTB2240配合使用,在同一批样本上同时检测多个脂代谢指标,减少批次间差异,让不同指标之间的数据具有直接的可比性。
方法的可重复性和发表可信度
一个检测方法能不能支撑文章发表,除了数据本身的质量,方法学的可信度也是审稿人会评估的维度。
比色法检测NADPH消耗在酶学领域有几十年的使用历史,方法原理和计算逻辑在文献中有充分的文档记录,不是一个需要向审稿人解释的新方法。基于这个原理开发的商品化试剂盒,如果有已发表文献的引用记录,在方法部分的描述和引用上就有据可查。
KTB2240(CheKine™脂肪酸合成酶活性检测试剂盒,微量法)目前已经出现在发表于Leukemia、PLoS Pathogens等期刊的文章中,覆盖肿瘤、神经炎症、代谢疾病、病毒感染和环境毒理多个研究方向。在方法部分引用这款试剂盒时,审稿人能通过已发表文献确认方法的使用记录,这对于缩短方法学审查时间、减少审稿人对方法可靠性的质疑,是有实际帮助的。
最后
FAS活性的比色法检测,从原理来看是一个思路非常直接的方法:让FAS在底物充足的环境里工作,测它消耗NADPH的速度,用NADPH在340 nm处的光学特性把消耗量转化为可以读取的吸光度变化。整个逻辑链条没有多余的环节,信号来源清晰,计算方式透明。
这个方法能成为主流,不是因为它在所有维度上都是最好的选择,而是因为它在灵敏度足够、操作简便、可以高通量、成本合理这几个对于大多数实验室来说最重要的维度上,给出了合理的平衡。
理解了方法背后的原理和逻辑,才能知道什么时候它是合适的选择,什么时候它的边界会被触及,以及在遇到数据问题的时候,应该从哪个方向去排查。这比单纯地会按照说明书操作,要有价值得多。
