从样本制备到结果计算:NO一氧化氮检测完整实验操作指南
拿到试剂盒,说明书翻开,步骤看起来不复杂。但真正上手做的时候,总有一些地方说明书写得很简略,或者几个步骤之间的逻辑不太清楚,结果卡在某个细节上。
这篇把完整流程从头捋一遍,重点说那些说明书里一笔带过、但实际操作里容易出问题的地方。
注意:本文所有参数(工作液比例、孵育条件、脱蛋白系数等)均基于 Abbkine KTB1400 的配方体系。如果你用的是其他品牌的NO检测试剂盒,请以该产品说明书为准,不要直接套用这里的数值。
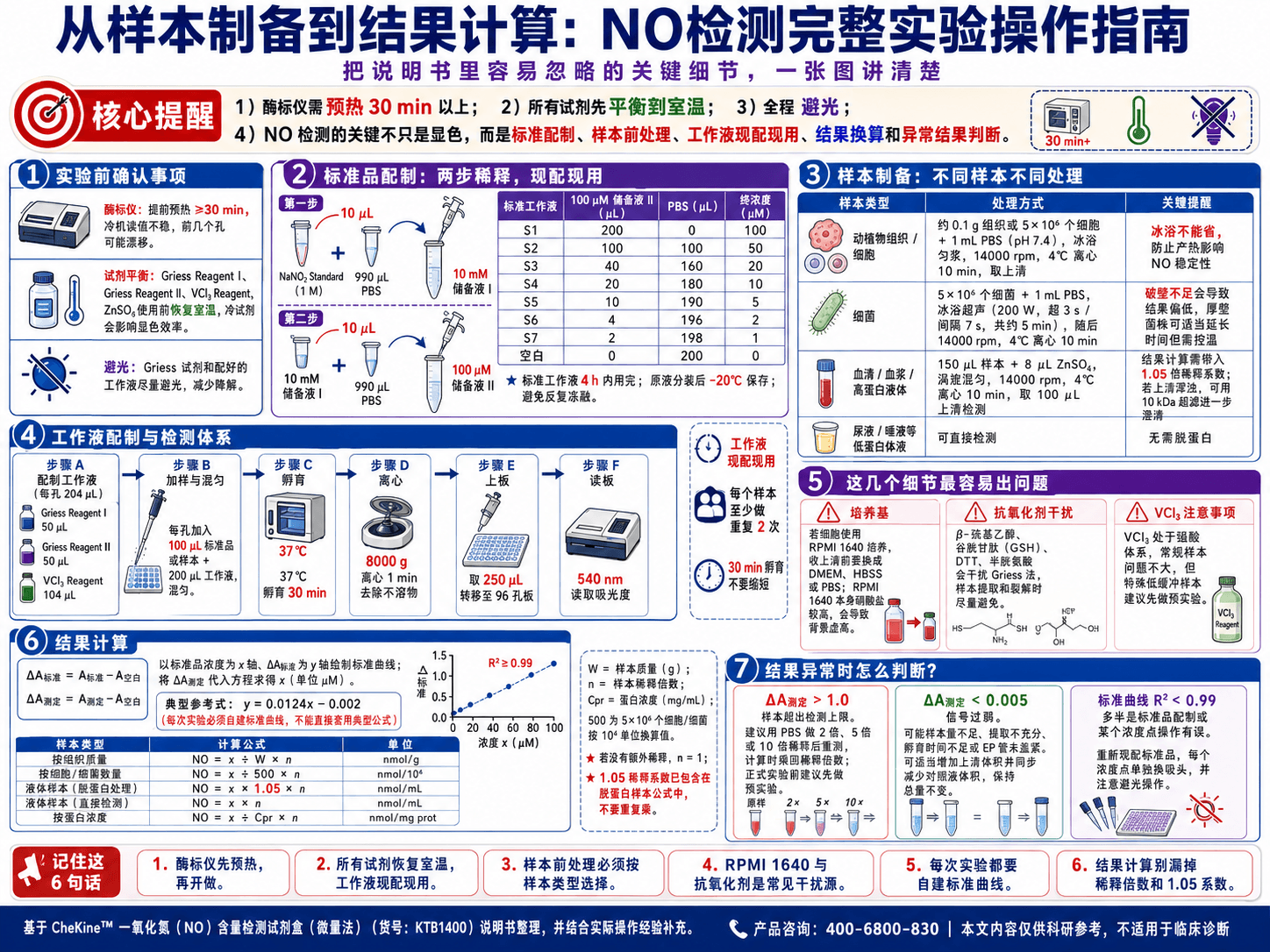
做实验之前,先把这几件事确认好
酶标仪要提前预热。 说明书写的是预热30分钟以上,这步不要省。酶标仪冷机状态下读值不稳定,前几个孔的数据会有漂移。预热这段时间正好用来配标准品和工作液。
所有试剂使用前平衡到室温。 Griess Reagent I、Griess Reagent II、VCl₃ Reagent、ZnSO₄都是即用型,从冰箱取出来之后不要直接用,先放到室温平衡。冷的试剂加进去会影响显色反应的效率。
全程避光。 Griess试剂见光会降解,操作过程中尽量不要让试剂长时间暴露在强光下,配好的工作液尤其要注意。
标准品配制:两步稀释,现配现用
标准品配制是两步稀释,很多人第一次做容易搞混。
第一步:取10 µL NaNO₂ Standard(原液浓度1 M),加990 µL PBS(pH 7.4),得到10 mM储备液I。
第二步:取10 µL 10 mM储备液I,加990 µL PBS,得到100 µM储备液II。后续的标准工作液都从这个100 µM储备液稀释出来。
| 标准工作液 | 100 µM储备液II(µL) | PBS(µL) | 终浓度(µM) |
|---|---|---|---|
| S1 | 200 | 0 | 100 |
| S2 | 100 | 100 | 50 |
| S3 | 40 | 160 | 20 |
| S4 | 20 | 180 | 10 |
| S5 | 10 | 190 | 5 |
| S6 | 4 | 196 | 2 |
| S7 | 2 | 198 | 1 |
| 空白 | 0 | 200 | 0 |
稀释后的标准工作液不稳定,必须在4小时内用完,不能留到下次。原液分装之后-20℃保存,每次取一管现配,不要反复冻融。
样本制备:不同样本处理方式不一样
动植物组织和细胞
称取约0.1 g组织,或者收集5×10⁶个细胞,加1 mL PBS(pH 7.4),冰浴匀浆。匀浆之后14000 rpm、4℃离心10分钟,取上清备用。
冰浴这个条件不能省,组织在匀浆过程中会产热,温度升高会加速NO的进一步氧化和降解,影响最终结果。
细菌
5×10⁶个细菌加1 mL PBS,冰浴超声破碎。超声条件:功率200 W,超声3秒、间隔7秒,重复30次,总计约5分钟。之后同样14000 rpm、4℃离心10分钟取上清。
超声参数对细菌破壁效率影响很大,功率不够或者次数不足,PRO出不来,数据会系统性偏低。如果你的菌株壁比较厚(比如某些革兰氏阳性菌),可以适当延长超声时间,但注意全程冰浴控温。
血清、血浆和高蛋白液体样本
这类样本需要脱蛋白之后再检测。流程:
150 µL样本加8 µL ZnSO₄,涡旋混匀,14000 rpm、4℃离心10分钟,取100 µL上清用于检测。
有几个细节:
第一,脱蛋白完成后上清如果还是浑浊的,说明蛋白没有完全沉淀。肝脏、脑组织这类富含脂质和复杂蛋白的样本,ZnSO₄有时候沉淀不彻底,浑浊的上清会干扰540 nm的读值,这种情况可以考虑用超滤管(截留分子量10 kDa)过滤一次,能有效降低背景。
第二,脱蛋白这一步引入了约1.05倍的稀释,计算结果时要乘上这个系数,不然数据会偏低。
尿液、唾液等低蛋白体液
直接取样检测,不需要脱蛋白步骤。
工作液配制和检测体系搭建
工作液按每孔200 µL配制,但建议按204 µL每孔来算,多配一点留出损耗:
- Griess Reagent I:50 µL
- Griess Reagent II:50 µL
- VCl₃ Reagent:104 µL
三个试剂混合即为工作液,现配现用,配好之后尽快用完,不要存放。
检测体系搭建:取100 µL稀释好的标准品或样本,加入200 µL工作液,混匀。每个样本建议至少重复两次。
混匀之后在37℃孵育30分钟。孵育过程中VCl₃完成NO₃⁻到NO₂⁻的还原,Griess试剂完成显色反应,这30分钟不要缩短。
孵育结束后,8000 g离心1分钟去除不溶物,从每个反应管取250 µL转移到96孔板,在540 nm读取吸光度。
有几个操作细节值得单独说
关于培养基:如果你的细胞样本用RPMI 1640培养,收上清之前一定要换掉培养基,换成DMEM、HBSS或者PBS之类的低硝酸盐培养液。RPMI 1640本身含有较高浓度硝酸盐,直接测背景会虚高,实验组和对照组的差异全被掩盖掉。
关于抗氧化剂:如果样本制备过程中用到了β-巯基乙醇、谷胱甘肽、DTT或者半胱氨酸,这些物质会干扰Griess法的显色反应,测出来的结果不可信。提取和裂解步骤里尽量避开这些试剂,实在无法避开的话,需要换其他方法来测NO。
关于VCl₃:VCl₃溶解在强酸溶液里,加进反应体系之后整体偏酸性。对大多数常规样本没有影响,但如果你的样本缓冲能力很弱,或者是某些特殊来源的生物液体,建议先跑一个预实验确认一下。
结果计算
先算ΔA值:
- ΔA标准 = A标准 - A空白
- ΔA测定 = A测定 - A空白
以标准品浓度为x轴,ΔA标准为y轴绘制标准曲线,把ΔA测定代入方程,得到x值(单位µM)。
典型标准曲线公式参考:y = 0.0124x - 0.002,但每次实验要自己做曲线,不能直接套这个数。
算出x之后,根据样本类型选对应的公式:
| 样本类型 | 计算公式 | 单位 |
|---|---|---|
| 按组织质量 | NO = x ÷ W × n | nmol/g |
| 按细胞/细菌数量 | NO = x ÷ 500 × n | nmol/10⁴ |
| 液体样本(脱蛋白处理) | NO = x × 1.05 × n | nmol/mL |
| 液体样本(直接检测) | NO = x × n | nmol/mL |
| 按蛋白浓度 | NO = x ÷ Cpr × n | nmol/mg prot |
变量说明:W = 样本质量(g);n = 样本稀释倍数;Cpr = 样本蛋白质浓度(mg/mL);500 = 细胞或细菌数量5×10⁶以10⁴为单位的换算值。
公式里的变量不多,但含义容易搞混,尤其是n(稀释倍数)。如果你的样本在制备过程中做过稀释,这里要乘进去;如果没有稀释,n = 1。脱蛋白那步引入的1.05倍稀释已经直接写进公式里了,不需要另外乘。
数据出来了,但看起来不对
ΔA测定大于1.0:样本NO含量超出检测上限了。用PBS按2倍、5倍或10倍稀释之后重新检测,计算时乘上对应的稀释倍数。建议在正式实验前先做一组预实验确认稀释比例,避免浪费试剂。
ΔA测定小于0.005:信号太弱,大概率是样本量不够,或者提取步骤有问题。可以适当增加加入反应体系的样本上清体积(同步减少对照液体积保持总量不变)。同时检查一下匀浆或超声是否充分,孵育时间是否足够,EP管在孵育过程中有没有盖紧。
标准曲线线性很差(R²低于0.99):基本上是标准品配制出了问题,或者某个浓度点操作有误。重新配标准品做一次,每个浓度点单独换吸头,操作过程注意避光。
以上操作流程基于 CheKine™ 一氧化氮(NO)含量检测试剂盒(微量法)(货号:KTB1400)说明书整理,结合实际操作经验补充了部分细节说明。产品咨询:400-6800-830 | 本文内容仅供科研参考,不适用于临床诊断
