NAD(H)和NADP(H)检测原理是什么?酶循环比色法详解
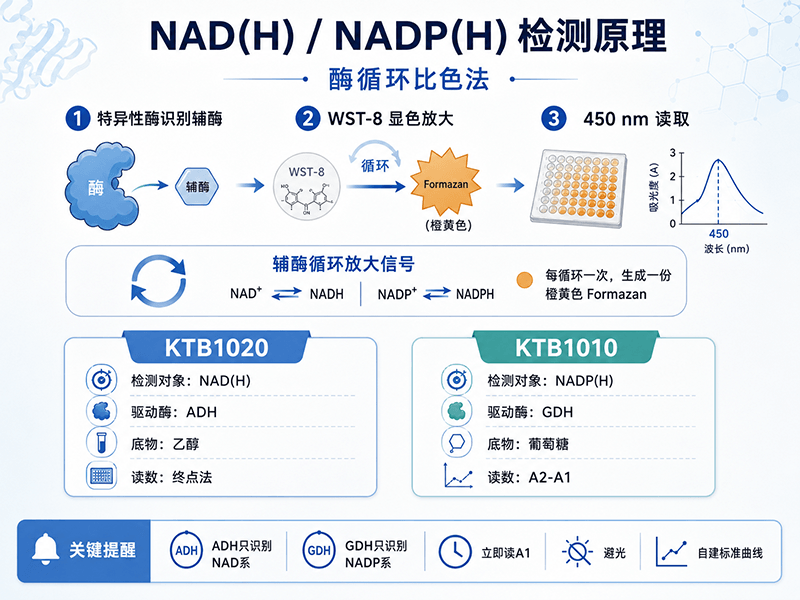
用比色法检测NAD(H)或NADP(H),操作上并不复杂——加试剂、孵育、读吸光值。但如果不理解背后的反应逻辑,实验中一旦出现异常,就很难判断问题出在哪一步。这篇文章从原理层面把酶循环比色法拆开来讲,读完之后你会明白每个试剂组分存在的理由,以及哪些操作细节是由原理决定的、不能随意省略的。
检测的核心难题:如何在复杂样本里只看到目标辅酶?
在讲具体反应之前,先说清楚这类检测面对的根本挑战。
细胞裂解液是一个极其复杂的混合物,里面有蛋白质、核酸、各种代谢物,还有大量在化学性质上类似的小分子。NAD(H)和NADP(H)在结构上高度相似,很多检测手段无法区分;更麻烦的是,NADH和NADPH都有天然荧光(激发波长约340 nm),理论上可以用荧光直接检测,但样本里其他荧光物质的干扰会让结果难以准确定量。
酶循环比色法解决这个问题的思路是:用高度特异的酶来"认人",用显色反应来"出结果"。酶的底物特异性替代了化学分离的步骤,显色反应把信号放大到可以用普通酶标仪在450 nm处定量读取。这套方案不需要荧光仪、不需要HPLC,普通实验室标配的96孔酶标仪就能完成检测,这是它在科研场景中被广泛使用的原因。
酶循环反应的基本逻辑
两款试剂盒(KTB1020检测NAD/H,KTB1010检测NADP/H)都基于同一套底层逻辑,只是用了不同的酶和底物。
核心思路是让目标辅酶在一个循环反应里被反复使用,每转一圈就产生一定量的显色信号,循环转得越多、信号积累得越强,最终信号强度与样品里目标辅酶的总量成正比。
这个设计有一个关键优势:辅酶本身不被消耗,它在氧化型和还原型之间反复转换,相当于一个信号放大器。样品里哪怕只有很少量的辅酶,也能通过多轮循环产生可检测的信号积累。这就是为什么这类试剂盒能在µM量级的浓度下实现可靠检测。
KTB1020的反应体系:乙醇脱氢酶驱动的NAD循环
反应过程
KTB1020(辅酶Ⅰ NAD(H)试剂盒)的工作液里包含NAD Cycling Enzyme Mix(以乙醇脱氢酶为核心)和EtOH Solution(乙醇底物),反应分两步走:
第一步(驱动循环):乙醇脱氢酶催化乙醇氧化,以NAD⁺作为电子受体,生成NADH。
乙醇 + NAD⁺ ──ADH──> 乙醛 + NADH + H⁺
第二步(产生信号):生成的NADH把WST-8(一种水溶性四唑盐)还原,生成橙黄色的formazan(甲臜),在450 nm处有最大吸收峰。
NADH + WST-8 ──Enhancer──> NAD⁺ + Formazan(橙黄色)
注意第二步里NAD⁺被再生了——它又可以回到第一步继续被乙醇脱氢酶还原为NADH。这就是"循环"的来源:NAD⁺和NADH在两步反应之间反复转换,每转一圈消耗一个WST-8分子、产生一个formazan分子,信号持续积累。
Enhancer(增强剂)的作用是加速NADH向WST-8的电子转移,没有它,这一步的反应速率会很慢,30 min的孵育时间内信号积累不足。
特异性从哪里来?
乙醇脱氢酶只能识别NAD⁺/NADH,不能识别NADP⁺/NADPH。这是酶的底物结合口袋决定的——NADP⁺比NAD⁺多一个2'-磷酸基团,而乙醇脱氢酶的活性位点没有容纳这个磷酸基团的空间。因此,样品里即使同时存在NAD(H)和NADP(H),这套反应体系只会"看到"NAD系辅酶,NADP系辅酶完全不参与循环,对最终读数没有贡献。
这也解释了为什么KTB1020和KTB1010可以在同一批样品里平行使用,结果互不干扰。
为什么NAD和NADH的标准曲线是同一条?
这是一个很多初次使用者会困惑的问题。
试剂盒只提供NAD标准品,而说明书里注明"NAD和NADH的标准曲线是相同的"。背后的逻辑是:在酶循环体系里,NAD⁺会立即被还原为NADH,NADH又会被WST-8氧化回NAD⁺,两者处于动态平衡。系统检测的是参与循环的辅酶总摩尔数,而不区分当下是氧化型还是还原型。等量摩尔的NAD⁺和NADH进入同一个反应体系,最终产生的formazan量是相同的,所以用一条标准曲线校准就足够了。
KTB1010的反应体系:葡萄糖脱氢酶驱动的NADP循环
反应过程
KTB1010(辅酶Ⅱ NADP(H)试剂盒)的工作液里包含NADP Cycling Enzyme Mix(以葡萄糖脱氢酶为核心)和Glucose(葡萄糖底物),反应同样分两步:
第一步(驱动循环):葡萄糖脱氢酶催化葡萄糖氧化,以NADP⁺作为电子受体,生成NADPH。
葡萄糖 + NADP⁺ ──GDH──> 葡萄糖酸 + NADPH + H⁺
第二步(产生信号):生成的NADPH还原WST-8,生成formazan,同时NADP⁺被再生回到第一步。
NADPH + WST-8 ──Enhancer──> NADP⁺ + Formazan(橙黄色)
显色原理与KTB1020完全相同,同样在450 nm处读取。葡萄糖脱氢酶只识别NADP系辅酶,NAD(H)不参与这套循环。
KTB1010为什么需要读两次?
这是两款试剂盒在操作上最明显的区别,值得从原理上说清楚。
KTB1020采用的是终点法:反应完成后(孵育30 min)读一次450 nm吸光值,减去空白孔背景,得到最终信号。
KTB1010采用的是动力学差值法:先在加入工作液后立即读一次A1,孵育30 min后再读一次A2,用ΔA=A2-A1作为定量信号。
为什么NADP/H的检测需要两次读值?葡萄糖脱氢酶体系在反应初始阶段的本底信号相对较高,单纯用终点值减去空白孔的校正精度不够,用动力学差值(反应前后的吸光度变化量)能更准确地捕捉因样品中NADP/H驱动产生的formazan增量,扣除试剂本身带来的背景漂移。
操作上的含义是:加入样品和工作液充分混匀后,必须立即读第一次,不能先做别的事再回来读。第一次读数拖延会导致反应已经进行了一部分,A1偏高,最终ΔA偏小,数据系统性偏低。
两套体系的对比
| KTB1020(NAD/H) | KTB1010(NADP/H) | |
|---|---|---|
| 驱动酶 | 乙醇脱氢酶(ADH) | 葡萄糖脱氢酶(GDH) |
| 循环底物 | 乙醇(EtOH) | 葡萄糖(Glucose) |
| 特异性根源 | ADH不识别NADP系 | GDH不识别NAD系 |
| 显色试剂 | WST-8 + Enhancer | WST-8 + Enhancer |
| 检测波长 | 450 nm | 450 nm |
| 读数方式 | 终点法(单次读数) | 动力学差值法(A2-A1) |
| 标准品 | 即用型液体NAD | 冻干粉NADPH,临用前复溶 |
两套系统用了相同的显色机制(WST-8/formazan/450 nm),但驱动循环的酶和底物完全不同——这个差异是两款产品能够分别专一检测各自靶标、互不干扰的根本原因。
标准曲线:为什么强烈建议每次实验自建?
试剂盒说明书里提供了典型标准曲线公式:
- KTB1020(NAD/H):y = 30.348x - 0.3208,R² = 0.9978
- KTB1010(NADP/H):y = 26.294x + 0.0516,R² = 0.9998
这两条曲线看起来线性很好,为什么说明书还是建议自建标准曲线,而不是直接套公式?
原因有几个。第一,酶循环反应的速率对温度敏感,不同实验室的室温条件、酶标仪预热状态、孵育时间的细微差异都会影响formazan的生成量,导致同样浓度的标准品在不同批次里产生略有差异的吸光值。第二,试剂盒的不同批号之间,酶活性可能存在正常范围内的波动。第三,标准曲线的斜率和截距会随上述因素漂移,用固定公式计算会引入系统误差。
自建标准曲线的操作成本很低——每次实验做一列标准孔,7个浓度梯度,1个空白,占用96孔板上不到一列的位置。这个代价换来的是当次实验条件下最准确的定量结果,是值得的。
如果由于样本数量确实排不下标准孔,才考虑参考说明书里的典型曲线——但需要在文章方法描述里如实写明。
这套方法检测的是什么,不是什么?
有一点在使用这类试剂盒时需要清楚认识,也是对数据解读很重要的前提。
酶循环比色法提取并检测的是样品中NAD(H)或NADP(H)的总量,包括游离态和蛋白结合态。而生理学研究中有时关注的"游离NAD⁺/NADH比值"——即细胞内真正处于游离状态、能参与酶催化的那部分辅酶的比例——是一个不同的概念。
游离辅酶的比值通常通过代谢物比值间接推算(比如用乳酸/丙酮酸比值推算细胞质游离NAD⁺/NADH),而非直接测量。这是因为提取过程本身会破坏蛋白-辅酶结合,导致提取后测到的总量里包含了从蛋白上释放出来的那部分。
这不是试剂盒的缺陷,而是所有基于提取的生化检测方法共同面对的限制。在方法描述和数据解读时,用"总NAD⁺/NADH含量"或"提取物中的NAD⁺/NADH水平"来表述,比"游离NAD⁺/NADH比值"更准确。同样地,如果审稿人追问游离辅酶的数据,直接测量的比色法无法给出这个答案,需要补充间接推算或采用其他检测手段。
提前了解这一点,能帮助你在实验设计阶段就想清楚数据能支撑什么结论,避免投稿时被动。
从原理到操作:哪些细节是由原理决定的?
理解了上面的反应机制,实验里有几个操作要求就不再是"说明书让这么做就这么做",而是有了具体的理由。
但在列这些细节之前,有一个前提必须先说清楚:NAD(H)和NADP(H)的提取过程本身就是整个实验链条里最脆弱的环节,操作速度和温度控制在这一步的重要性,远超后续的检测体系配制。
辅酶对温度极度敏感——即便是在酸性或碱性保护环境下,样本温度升高也会加速降解。提取的正确流程是:匀浆或超声破碎全程在冰上进行,破碎完成后立即加入提取缓冲液,加热煮沸步骤按说明书时间严格控制(5 min,不要延长),煮沸结束后立即放回冰上等待中和离心。从样本离开低温环境到加入提取液之间的时间窗口,能压缩到多短就压缩到多短。
组织样本的前处理尤其需要注意:剪碎组织这一步如果在室温下慢慢操作,辅酶的降解已经开始了,后面提取再标准也弥补不了。建议的做法是取材后立即液氮淬灭,在冷冻状态下称重,然后直接转入预冷的匀浆体系,不给样本在室温下停留的机会。
这个前提说清楚之后,再来看检测体系里其他几个由原理决定的操作要求:
工作液必须现配现用,加入后立即混匀。 酶循环反应一旦启动就开始产生formazan,如果加入工作液后有延迟,各孔的反应时间不一致,读数会出现孔间差异。说明书特别强调"工作液加入需要迅速,各组分需要彻底混合",原因就在这里。
WST-8和Enhancer必须避光保存,实验过程中冰上避光放置。 WST-8是光敏性化合物,光照会导致非酶催化的自发还原,产生本底formazan信号,抬高背景。实验台上不要让这两个组分长时间暴露在光线下。
KTB1010加入样品后必须立即读A1。 前面解释过原因:拖延读第一个时间点会导致ΔA系统性偏低。这个步骤不能和其他操作穿插进行。
提取缓冲液不能加错。 酸性提取液(NAD/NADP Extraction Buffer)专门用来提取氧化型,碱性提取液(NADH/NADPH Extraction Buffer)专门用来提取还原型。加错会在样本制备阶段就把目标辅酶降解掉,后续检测到的信号接近零,不是试剂盒失效,是提取这步已经把样本毁了。
孵育30 min,不要随意缩短。 酶循环需要足够的时间积累formazan信号,尤其是样本浓度较低时,缩短孵育时间会导致信号积累不足,读数偏低。如果实验时间紧张,可以考虑提前规划操作流程,而不是缩短孵育时间。
小结
NAD(H)和NADP(H)的酶循环比色法,核心是用特异性酶驱动辅酶在氧化型和还原型之间循环,每转一圈产生一个formazan分子,累积的显色信号与样品中的辅酶总量成正比。KTB1020依赖乙醇脱氢酶和乙醇底物检测NAD系,KTB1010依赖葡萄糖脱氢酶和葡萄糖底物检测NADP系,两套系统在酶的底物特异性上完全隔离,可以平行使用。
两款产品都能同时检测氧化型和还原型,最终给出各自的含量及比值,用于反映细胞的氧化还原状态。方法检测的是提取物中的总辅酶量(游离+蛋白结合态),这一点在数据解读时需要准确表述。
如果你已经看完了这篇原理介绍,下一步可以参考KTB1020和KTB1010各自的实验操作详解,按样本类型选择对应的提取方案,完成从样本制备到结果计算的完整流程。
